Documentation
Contents
- Abbreviations

- Populations
- Diseases
- Variant and Annotation Data
- Population and Disease-Associated Variant Data Collection
- Saturation Mutagenesis
- Pre-computed Variant Pathogenicity Predictions
- AlphaFold2 and AlphaMissense
- Protein Sequence Data
- Definition of Titin Domain Boundaries
- Mapping of Titin Isoforms
- Annotation of Shared Structural Positions
- Protein Structural Data
- Protein Data Bank Experimental Structures
- Homology Modelling of Titin Domains
- AlphaFold2 Predicted Structures
- Definition of Structural Elements
- Protein-Protein Interactions and Site Annotations
- TITINdb2 Variant Converter
- Citing TITINdb2
- References
Link types
The different links in TITINdb2 use the following convention:
- Internal links to TITINdb2:
- External links to other websites:

- Download links:
Abbreviations
Populations
| AFR | African/African American |
| AMR | Latino |
| AMS | Amish |
| ASJ | Ashkenazi Jewish |
| EAS | East Asian |
| EUR | European (1000 genomes only) |
| FIN | Finnish |
| MDE | Middle Eastern |
| NFE | Non-Finnish European |
| SAS | South Asian |
| OTH | Other |
N.B. if a population classification is not used by a database the value None is given.
Diseases
| CM | Cardiomyopathy (any) |
| DCM | Dilated Cardiomyopathy |
| HCM | Hypertrophic Cardiomyopathy |
| ARVC | Arrythmogenic Right Ventricular Cardiomyopathy |
| RCM | Restrictive Cardiomyopathy |
| MD | Muscle Disease |
| MmD | Multi-minicore Disease |
| MmD-HD | Multi-minicore Disease with Heart Disease |
| CNM | Centronuclear Myopathy |
| LGMD2J | Limb-Girdle Muscular Dystrophy Type 2J |
| TMD | Tibial Muscular Dystrophy |
| MFM | Myofibrillar Myopathy |
| HMERF | Hereditary Myopathy with Early Respiratory Failure |
| IH | Inguinal Hernia |
| T1P | Type 1 Fibre Predominance |
| CFTD | Congenital Fibre Type Disproportion |
| Other | Unknown or novel titinopathy |
Variant and Annotation Data
Population and Disease-Associated Variant Data Collection
Population missense variant data was originally obtained from 3 sources: the 1000 Genomes Project (1KGP) [1], gnomAD database 2.1.1 (gnomAD2) and gnomAD database 3.1.2 (gnomAD3) [2]. Data from 1KGP was obtained from dbSNP [3], while data from gnomAD2 and gnomAD3 were obtained from the gnomAD Broad Institute web server [4]. The gnomAD2 release is composed of 125,748 exomes and 15,708 genomes, while gnomAD3 covers 76,156 genomes. Data from 1KGP is present in gnomAD databases, and there is minor overlap between gnomAD2 and gnomAD3 genomes surveyed.
As of November 2023, gnomAD database 4.0.0 has been released, with a vast increase in both genome and variant quantity over both versions 2 and 3, composed of 730,947 exomes and 76,215 genomes. While all data from the versions 2.1.1 and 3.1.2 data sets are reported to be present in the version 4.0.0 release, there are a non-negligible number of variants in titin present in one or the other of these older releases that are not reported in the newest database. Consequently, TITINdb2 includes all three databases as separate sources of variation. Users should be aware that gnomAD database 4.0.0 (gnomAD4) possesses a much higher sampling of individuals with European ancestry compared to the older releases, thanks to the inclusion of data from the UK Biobank [5]; consequently, we recommend increased scrutiny of reported minor allele frequency (MAF) within this data set, and to be mindful of the calculated allele frequency in ethnic subpopulations (a breakdown of the genetic ancestry of all gnomAD data sets can be found in the gnomAD v4.0.0 announcement [6]).
Disease-associated titin nsSNVs were originally obtained from Chauveau et al. (2012) [7]. The current build of TITINdb2 includes all known reported disease-associated titin missense variants obtained from a search of the available literature up to the end of 2023; notably, this excludes certain variants reported in other databases of reported disease-associated variants, such as ClinVar [8], where no accompanying peer-reviewed literature in support is provided. The published form of this literature review includes additional information about each of the variants reported in TITINdb2 [9].
Saturation Mutagenesis
Saturation mutagenesis, or deep mutational scanning (DMS), refers to the systematic evaluation of the impacts of single protein residue alterations at all positions; in experimental protocols, this involves high-throughput assays assessing a measurable parameter for protein constructs, such as stability or binding affinity, for all possible single amino acid substitutions at all possible sequence positions in the protein [10]. Similarly, in silico prediction tools can be used to produce computational mutational scans, allowing for comparison of vast numbers of protein substitutions in large proteins such as titin. TITINdb2 incorporates saturation mutagenesis data from a number of different prediction tools, which may be freely accessed using the "SatMut SAVs" table on the domain structure page for any given domain.
Saturation mutagenesis pathogenicity predictions for all SAVs present in titin's canonical (N2AB) isoform were extracted from the pre-computed databases for PolyPhen-2 [11] using their online web server. These predictions were then used as input for saturation mutagenesis predictions for all N2AB SAVs located within domain boundaries for Rhapsody [12], a predictor which uses sequence-, structural-, and dynamics-based features to predict pathogenicity. As Rhapsody's web server does not have access to the full structural coverage of titin available in TITINdb2, we implemented a local pipeline incorporating pre-computed PolyPhen-2 predictions and titin domain models from AlphaFold2 (see below) to generate predictions. As in the previous build of TITINdb, all possible SAVs within domain boundaries in the inferred complete (IC) isoform of titin were assessed using mCSM [13] to predict protein stability by web server query, using homology models of titin domains generated by Modeller (see below). Protein stability predictions were also generated using DUET [14], for all possible nsSNVs only.
In addition to the generated saturation mutagenesis data, TITINdb2 also includes saturation mutagenesis predictions made available as pre-computed data for the community. Pathogenicity predictions for all possible nsSNVs in titin's canonical isoform are available for the meta-predictors Condel [15] and REVEL [16]; Condel predictions were obtained from FannsDB [17], while REVEL predictions were mapped from chromosomal to protein coordinates before being added to TITINdb2. Finally, the recent release of AlphaMissense [18] has included pre-computed predictions for all possible SAVs at every position in titin, both for the canonical N2AB isoform and for the inferred complete isoform. As these predictions contain minor differences, both are included in saturation mutagenesis data in TITINdb2 for the sake of comparison; both are sourced from the distribution of AlphaMissense predictions through Zenodo [19].
Pre-computed Variant Pathogenicity Predictions
Pathogenicity predictions for reported SNVs in population or pathogenic variant databases have been collected in various online repositories; we include predictions sourced from one of the most comprehensive of these databases, dbNSFPv4 [20], for nsSNVs reported therein. Of the predictors included in dbNSFPv4, we selected 13 pathogenicity prediction algorithms which were both commonly cited in comparisons of pathogenicity prediction accuracy and not sourced from other databases, and added these annotations to TITINdb2. A full list of all available variant impact prediction algorithms in TITINdb2, including both saturation mutagenesis and selected variant annotations, is given below:
| Predictor Name | Source | Description | Availability | Reference |
|---|---|---|---|---|
| AlphaMissense | Predictions for AlphaMissense (Zenodo) [19] | Pathogenicity predictor (large language model trained on human/primate variant frequency databases) | Saturation mutagenesis of SAVs (all positions) | Cheng et al. (2023) [18] |
| CADD | dbNSFPv4 [20] | Pathogenicity meta-predictor (sequence conservation, other predictors) | Selected nsSNVs | Rentzsch et al. (2019) [21] |
| Condel | FannsDB [17] | Pathogenicity meta-predictor (FATHMM, MutationAssessor) | All possible nsSNVs | González-Pérez & López-Bigas (2011) [15] |
| DUET | Calculation with homology models (web server) | Protein stability meta-predictor (mCSM, SDM) | All possible nsSNVs | Pires et al. (2014b) [14] |
| FATHMM | dbNSFPv4 | Pathogenicity predictor (sequence homology, disease weighting) | Selected nsSNVs | Shihab et al. (2013) [22] |
| M-CAP | dbNSFPv4 | Pathogenicity predictor (sequence homology, genetic constraints) | Selected nsSNVs | Jagadeesh et al. (2016) [23] |
| mCSM | Calculation with homology models (web server) | Protein stability predictor (graph-based structural signatures) | Saturation mutagenesis of SAVs (all domains) | Pires et al. (2014a) [13] |
| MetaSVM | dbNSFPv4 | Pathogenicity meta-predictor (11 other predictors, allele frequency) | Selected nsSNVs | Dong, C. et al. (2015) [24] |
| MetaLR | dbNSFPv4 | Pathogenicity meta-predictor (11 other predictors, allele frequency) | Selected nsSNVs | Dong, C. et al. (2015) [24] |
| MPC | dbNSFPv4 | Pathogenicity predictor (sequence homology, genetic constraints) | Selected nsSNVs | Samocha et al. (2017) [25] |
| MutationAssessor | dbNSFPv4 | Pathogenicity predictor (sequence homology) | Selected nsSNVs | Reva et al. (2011) [26] |
| MutationTaster | dbNSFPv4 | Pathogenicity predictor (sequence homology) | Selected nsSNVs | Schwarz et al. (2014) [27] |
| MutPred | dbNSFPv4 | Pathogenicity predictor (sequence homology, conservation, structural change) | Selected nsSNVs | Pejaver et al. (2020) [28] |
| MVP | dbNSFPv4 | Pathogenicity predictor (sequence conservation, structural features, other predictors) | Selected nsSNVs | Qi et al. (2021) [29] |
| PolyPhen-2 | Retrieval from web server | Pathogenicity predictor (sequence homology, structural annotation) | Saturation mutagenesis of SAVs (N2AB domains) | Adzhubei et al. (2013) [11] |
| PrimateAI | dbNSFPv4 | Pathogenicity predictor (deep learning from sequence alignments) | Selected nsSNVs | Sundaram et al. (2018) [30] |
| PROVEAN | dbNSFPv4 | Pathogenicity predictor (sequence homology) | Selected nsSNVs | Choi et al. (2012) [31] |
| REVEL | REVEL genome segment files [28] | Pathogenicity meta-predictor (13 other predictors) | All possible nsSNVs | Ioannidis et al. (2016) [16] |
| Rhapsody | Calculation with AlphaFold2 models (local pipeline) | Pathogenicity predictor (sequence conservation, structural features, coarse-grained dynamics) | Saturation mutagenesis of SAVs (N2AB domains) | Ponzoni et al. (2020) [12] |
| SIFT | dbNSFPv4 | Pathogenicity predictor (sequence conservation) | Selected nsSNVs | Ng & Henikoff (2003) [33] |
AlphaFold2 and AlphaMissense
Recent developments in computational prediction, driven by advances in artificial intelligence and large language models, have given researchers access to a wide variety of pre-computed data; chief among these are the protein structure prediction tool AlphaFold2 [34] and the previously mentioned variant pathogenicity prediction tool AlphaMissense [18], both developed by DeepMind. In addition to the methodologies, pre-computed data for both structural models of all proteins and predicted pathogenicity for all possible SAVs in the human proteome have been released; however, accessing the data for titin is not straightforward, owing to its immense size. Thus, in TITINdb2 both the AlphaFold2 structural models for titin domains and AlphaMissense predictions of pathogenicity for titin SAVs have been extracted from the proteome data files and are readily available alongside other predicted structures and variant impact scores. AlphaMissense predictions have been sourced from both canonical and isoform proteome data sets via Zenodo (see above); AlphaFold2 structures for both single and tandem titin domain constructs have been extracted from predicted proteome structures and can be viewed in the provided molecule viewer, while the source 1400-residue proteome structures can be downloaded from the structures page for a given protein domain (for details, see Protein Structure Data, below).
Protein Sequence Data
Definition of Titin Domain Boundaries
HMMER [35] was used to scan the protein sequence of the titin IC isoform (NP_001254479.2, obtained from the RefSeq database [36]) against Pfam seed libraries [37]. Where hits overlapped the hit with the lowest E-value was accepted. When the lowest E-value hit for a region was greater than 0.0001, additional evidence was required to accept a hit. This was the case for domain Ig-94, which was identified with an E-value higher than the threshold (0.0001), but is verified by PDB structures 1WAA, 1TIU and 1TIT. Ig-88, Ig-89, Ig-90 and Ig-98 were also identified with E-values higher than the threshold; however when the titin sequence was scanned using an HMM created from an alignment of all (165) other titin Ig domains, these were identified with high significance (5.9E-11, 1.2E-12, 2.9E-10,7.4E-12).
Sequence logos created using Weblogo [38] (see Fig 1) showing aligned titin Fn3 sequences, differ substantially from such logos depicting Pfam seed alignments, particularly towards the end of the sequence where the conservation drops off gradually. Therefore, the boundary does not appear to be clearly defined from sequence alone. When mapped onto structure it becomes clear that the Pfam-defined boundaries do not cover the whole domain. Due to this information it was decided the Pfam Fn3 domain boundaries were not appropriate. Therefore, Fn3 domains were initially identified using Pfam/HMMER and the sequences of these domains, including an extra 5 amino acids upstream and 16 amino acids downstream of the Pfam defined boundary, were aligned using T-Coffee [39]. This alignment was cut using structural information from available titin Fn3 crystal structures, in particular 3LPW. An HMM was created from this alignment and titin scanned again using this HMM to redefine Fn3 boundaries.
Mapping of Titin Isoforms
Isoform sequences were obtained from RefSeq [36]. Stretcher [40] was used to align all titin isoforms to the IC isoform. Positions were mapped according to these alignments.
| Isoform | Protein Length (aa) | UniProt ID | Ensembl Protein ID | NCBI RefSeq Protein ID |
|---|---|---|---|---|
| Inferred Complete (IC) | 35991 | Q8WZ42-12 |
ENSP00000467141.1 |
NP_001254479.2 |
| Canonical (N2AB) | 34350 | Q8WZ42-1 |
ENSP00000465570.1 |
NP_001243779.1 |
| Primary Cardiac (N2B) | 26926 | Q8WZ42-3 |
ENSP00000434586.1 |
NP_003310.4 |
| Primary Skeletal (N2A) | 33423 | Q8WZ42-11 |
ENSP00000343764.6 |
NP_596869.4 |
| Cardiac Novex-1 (novex1) | 27051 | Q8WZ42-10 |
ENSP00000352154.5 |
NP_597676.3 |
| Cardiac Novex-2 (novex2) | 27118 | NA | ENSP00000340554.6 |
NP_597681.4 |
| Cardiac Novex-3 (novex3) | 5604 | Q8WZ42-6 |
ENSP00000354117.4 |
NP_596870.2 |
Annotation of Shared Structural Positions
While there are limited numbers of variants reported at each individual position in titin, the Ig and Fn3 domains that make up the protein have a high sequence identity, and information about variation at a given position in a domain may be provided by variants at the same structural position in homologous domains. Multiple sequence alignments of all 169 Ig domains and 132 Fn3 domains in titin were constructed and aligned positions were allocated; the structural position annotation, accessible as a link from the page for any residue located within a domain, allows the user to navigate to a page where all reported variants at that shared structural position, as well as the wild-type reside and the domains they occur in, can be viewed.
Consensus structural alignments were generated using PROMALS3D [41] with available experimental X-ray crystal structures from PDBe (see below) for both Ig and Fn3 domains; these were then mapped back onto the multiple sequence alignments of all Ig and Fn3 domains, respectively, and shared structural positions were annotated with consensus secondary structure information at that position.
Protein Structure Data
Protein Data Bank Experimental Structures
The Protein Data Bank in Europe (PDBe) [42] was searched for experimental structures of single or tandem titin domain constructs, solved with X-ray crystallography, electron microscopy, or NMR spectroscopy. The following experimental structures are available in TITINdb2 for visualisation of variants on their respective domains:
| PDB ID | Reported Domain | UniProt Domain | TITINdb2 Domain | Chain | Solution Method | Resolution |
|---|---|---|---|---|---|---|
| 2F8V | Titin-Nterm | Ig-like 1 | Ig-1 | A | X-Ray Diffraction | 2.75Å |
| Ig-like 2 | Ig-2 | |||||
| 2A38 | Titin-Nterm | Ig-like 1 | Ig-1 | A | X-Ray Diffraction | 2.00Å |
| Ig-like 2 | Ig-2 | |||||
| 6FWX | Titin-Z1 | Ig-like 1 | Ig-1 | A | X-Ray Diffraction | 3.00Å |
| Titin-Z2 | Ig-like 2 | Ig-2 | ||||
| 6SDB | Titin-Z1 | Ig-like 1 | Ig-1 | A | X-Ray Diffraction | 2.80Å |
| Titin-Z2 | Ig-like 2 | Ig-2 | ||||
| 1YA5 | Titin-Z1 | Ig-like 1 | Ig-1 | A | X-Ray Diffraction | 2.44Å |
| Titin-Z2 | Ig-like 2 | Ig-2 | ||||
| 6DL4 | Titin-Z10 | Ig-like 9 | Ig-9 | A | Solution NMR | |
| 1G1C | Titin-I1 | Ig-like 10 | Ig-10 | A | X-Ray Diffraction | 2.10Å |
| 4QEG | Titin-I10 | Ig-like 16 | Ig-19 | A | X-Ray Diffraction | 2.00Å |
| 5JDJ | Titin-I10 | Ig-like 16 | Ig-19 | A | X-Ray Diffraction | 1.74Å |
| 5JDD | Titin-I9 | N/A | Ig-18 | A | X-Ray Diffraction | 1.53Å |
| Titin-I10 | Ig-like 16 | Ig-19 | ||||
| Titin-I11 | Ig-like 17 | Ig-20 | ||||
| 5JDE | Titin-I9 | N/A | Ig-18 | A | X-Ray Diffraction | 1.90Å |
| Titin-I10 | Ig-like 16 | Ig-19 | ||||
| Titin-I11 | Ig-like 17 | Ig-20 | ||||
| 2RIK | Titin I67 | Ig-like 64 | Ig-70 | A | X-Ray Diffraction | 1.60Å |
| Titin I68 | Ig-like 65 | Ig-71 | ||||
| Titin I69 | Ig-like 66 | Ig-72 | ||||
| 3B43 | Titin I65 | Ig-like 62 | Ig-68 | A | X-Ray Diffraction | 3.30Å |
| Titin I66 | Ig-like 63 | Ig-69 | ||||
| Titin I67 | Ig-like 64 | Ig-70 | ||||
| Titin I68 | Ig-like 65 | Ig-71 | ||||
| Titin I69 | Ig-like 66 | Ig-72 | ||||
| Titin I70 | Ig-like 67 | Ig-73 | ||||
| 5JOE | Titin I81 | N/A | Ig-84 | A | X-Ray Diffraction | 2.00Å |
| 7AHS | Titin-N2A Ig81 | Ig-like 77 | Ig-84 | A | X-Ray Diffraction | 2.04Å |
| Titin-N2A Ig82 | Ig-like 78 | Ig-85 | ||||
| Titin-N2A Ig83 | Ig-like 79 | Ig-86 | ||||
| 6YJ0 | Titin I83 | Ig-like 79 | Ig-86 | A | Solution NMR | |
| 1TIT | Titin-I27 | N/A | Ig-94 | A | Solution NMR | |
| 1TIU | Titin-I27 | N/A | Ig-94 | A | Solution NMR | |
| 1WAA | Titin-I27 | N/A | Ig-94 | A | X-Ray Diffraction | 1.80Å |
| 4O00 | Titin-A3 | FnIII 3 | Fn3-3 | A | X-Ray Diffraction | 1.85Å |
| 8BXR | Titin I109 | N/A | Ig-109 | A | X-Ray Diffraction | 2.70Å |
| Titin I110 | FnIII 4 | Fn3-4 | ||||
| Titin I111 | FnIII 5 | Fn3-5 | ||||
| 8BW6 | Titin I110 | FnIII 4 | Fn3-4 | A | X-Ray Diffraction | 1.95Å |
| 8BVO | Titin I110 | FnIII 4 | Fn3-4 | A | X-Ray Diffraction | 2.55Å |
| Titin I111 | FnIII 5 | Fn3-5 | ||||
| 1BPV | Titin-A71 | FnIII 62 | Fn3-62 | A | Solution NMR | |
| 3LPW | Titin-A77 | FnIII 66 | Fn3-66 | A | X-Ray Diffraction | 1.65Å |
| Titin-A78 | FnIII 67 | Fn3-67 | ||||
| 3LCY | Titin-A164 | Ig-like 140 | Ig-156 | A | X-Ray Diffraction | 2.50Å |
| Titin-A165 | N/A | Ig-157 | ||||
| 2J8H | Titin-A168 | Ig-like 141 | Ig-158 | A | X-Ray Diffraction | 1.99Å |
| Titin-A169 | Ig-like 142 | Ig-159 | ||||
| 2J8O | Titin-A168 | Ig-like 141 | Ig-158 | A | X-Ray Diffraction | 2.49Å |
| Titin-A169 | Ig-like 142 | Ig-159 | ||||
| 2ILL | Titin-A168 | Ig-like 141 | Ig-158 | A | X-Ray Diffraction | 2.20Å |
| Titin-A169 | Ig-like 142 | Ig-159 | ||||
| 2NZI | Titin-A168 | Ig-like 141 | Ig-158 | A | X-Ray Diffraction | 2.90Å |
| Titin-A169 | Ig-like 142 | Ig-159 | ||||
| Titin-A170 | FnIII 132 | Fn3-132 | ||||
| 4JNW | Titin Kinase | Protein kinase | Kinase-1 | A | X-Ray Diffraction | 2.06Å |
| 1TKI | Titin Kinase | Protein kinase | Kinase-1 | A | X-Ray Diffraction | 2.00Å |
| 6YGN | Titin-A168 | FnIII 132 | Fn3-132 | A | X-Ray Diffraction | 2.40Å |
| Titin Kinase | Protein kinase | Kinase-1 | ||||
| Titin-M1 | Ig-like 143 | Ig-160 | ||||
| 2BK8 | Titin-M1 | Ig-like 143 | Ig-160 | A | X-Ray Diffraction | 1.69Å |
| 6HCI | Titin-M3 | Ig-like 145 | Ig-162 | A | X-Ray Diffraction | 2.12Å |
| 6H4L | Titin-M4 | Ig-like 146 | Ig-163 | A | X-Ray Diffraction | 1.60Å |
| 3QP3 | Titin-M4 | Ig-like 146 | Ig-163 | A | X-Ray Diffraction | 2.00Å |
| 1NCT | Titin-M5 | Ig-like 147 | Ig-164 | A | Solution NMR | |
| 1NCU | Titin-M5 | Ig-like 147 | Ig-164 | A | Solution NMR | |
| 1TNN | Titin-??? | Ig-like 147 | Ig-164 | A | Solution NMR | |
| 1TNM | Titin-??? | Ig-like 147 | Ig-164 | A | Solution NMR | |
| 3PUC | Titin-M7 | Ig-like 149 | Ig-166 | A | X-Ray Diffraction | 0.96Å |
| 4C4K | Titin-M10 | Ig-like 152 | Ig-169 | T | X-Ray Diffraction | 1.95Å |
| 4UOW | Titin-M10 | Ig-like 152 | Ig-169 | 1 | X-Ray Diffraction | 3.30Å |
| 3Q5O | Titin-M10 | Ig-like 152 | Ig-169 | A | X-Ray Diffraction | 2.05Å |
| 2Y9R | Titin-M10 | Ig-like 152 | Ig-169 | T | X-Ray Diffraction | 1.90Å |
| 2WP3 | Titin-M10 | Ig-like 152 | Ig-169 | T | X-Ray Diffraction | 1.48Å |
| 2WWK | Titin-M10 | Ig-like 152 | Ig-169 | T | X-Ray Diffraction | 1.70Å |
| 2WWM | Titin-M10 | Ig-like 152 | Ig-169 | D | X-Ray Diffraction | 2.30Å |
| 3KNB | Titin-Cterm | Ig-like 152 | Ig-169 | A | X-Ray Diffraction | 1.40Å |
All of the above structures represent a complete domain or domains aligning to H. sapiens titin; incomplete structures or epitopes are excluded, as well as titin domains from non-human model organisms. Additionally, two human titin structures available in PDB are not present in TITINdb2: 6I0Y is a large TnaC-stalled ribosome complex to which titin Ig-94 is bound; 2RQ8 is a mutant domain Ig-94, which includes variant Y14326P for analysis of domain destabilisation and thus does not align to the domain sequence.
Homology Modelling of Titin Domains
Prior to the proliferation of Deep Learning-based protein structure prediction algorithms, visualisation of individual titin domains lacking reliable, experimentally solved structures was achieved through the use of template modelling approaches. In the first version of this database, an automated homology modelling pipeline was set up to provide structural coverage to domains lacking solved experimental structures. The pipeline takes a FASTA file of domain sequences as input and uses PDB structures publicly available prior to March 2017 as templates. The overall modelling process can be seen in Fig 2A and a flow diagram detailing the template selection process is depicted in Fig 2B. The template search, modelling and model assessment were performed using Modeller [43], the alignment of query and templates performed using 3DCoffee [39], and the overall pipeline written in Python 2.7.

The I-TASSER server [44] was used to model Ig-112 as a satisfactory (negative) zDOPE score could not be obtained using Modeller.
AlphaFold2 Predicted Structures
Template-based models in TITINdb2 have been supplemented by models predicted by newer Deep Learning-based approaches, namely AlphaFold2 [34]. AlphaFold has provided the structural biology community with predictions of all proteins in the human proteome via their online Protein Structure Database [45]; however, owing to the size of titin, its structure cannot be predicted as a single protein. Thus, we obtained the full proteome predictions from the database and extracted the predictions that covered the genetic location of the titin gene. This is made up of 166 overlapping "fragments" of 1400 residues each, which together provide redundant structural coverage across the entirety of the titin protein. Each individual domain is typically represented in 6-7 fragments; these were extracted using the domain boundaries defined above, and re-numbered using PyMol [46], to produce an ensemble of models for both single and two-domain tandem systems (see Fig. 3).

The original AlphaFold2 fragments are named sequentially; for example, the fragment AF-Q8WZ42-F5-model_v1
is the fifth fragment in the proteome data, and covers residues 801-2200 of the canonical sequence of titin. The subsequent
fragment, AF-Q8WZ42-F6-model_v1, the sixth fragment, covers residues 1001-2400 of N2AB titin. This
continues until the final fragment is reached, AF-Q8WZ42-F166-model_v1, which covers residues
33001-34350. This naming convention continues for the extracted models of domain constructs; for example, the
model TTN-IC_Ig-3_AF-Q8WZ42-F5-model_v1.pdb is the single domain Ig-3 (IC isoform) extracted from
the fifth fragment of titin in AlphaFoldDB.
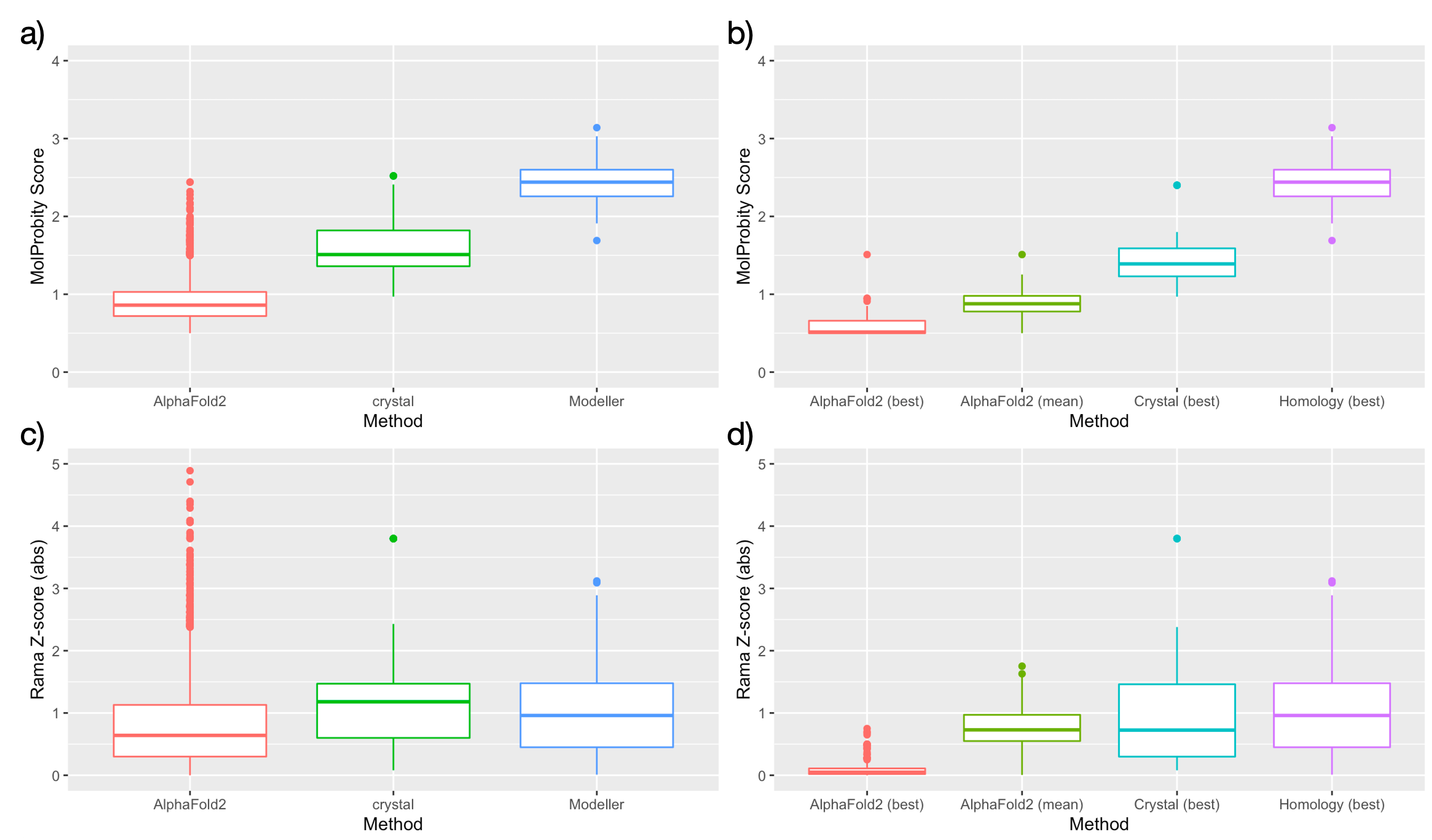
To compare the relative quality of models from Modeller and AlphaFold2, we used a local version of MolProbity [47] to compute summary statistics for each individual model. Two metrics are reported for all models: MolProbity score, an estimate of "effective resolution" calculated from rotamer, Ramachandran, and clash scores; and Rama Z-score, a single-value summary of Ramachandran favoured/outlier residues. Available structures for each domain may be sorted by either of these metrics. AlphaFold2 models were overall estimated to be of substantially higher quality on average than Modeller homology models by MolProbity score, and the best-ranked AlphaFold2 models for each domain compare favourably to estimates of available crystal structures. However, there is little difference between models estimated by Rama Z-score, and almost all models are within accepted limits for model quality (absolute Rama Z-score < 2.0).
As the canonical sequence was used by AlphaFold2 to model all fragments, there is no coverage of the domains located in exons 46, 47 and 48 of the TTN gene. These correspond to Ig-25 (IC, novex-1), Ig-26 (IC, novex-2), and the alternative novex-3 domains (denoted in TITINdb2 as Ig-25_novex3, Ig-26_novex3, Ig-27_novex3, Ig-28_novex3, Ig-29_novex3, and Ig-30_novex3). To remedy this, we used an adaptation of the AlphaFold2 pipeline distributed through Google Colaboratory, ColabFold [48], which allows for remote access to GPUs for prediction, and incorporates a faster but less exhaustive multiple sequence alignment search. The models produced are also not relaxed by the Amber force field because of memory limitations. The resulting ColabFold structures are of lower general quality than the AlphaFold2 predicted structures, but are still of high overall quality compared to template-based models.
Definition of Structural Elements
Residues in surface and core regions were defined using POPS [49]. Residues with a Q(SASA) (quotient solvent accessible surface area) > 0.3 were defined as being surface residues and those with a Q(SASA) ≤ 0.3 defined as core residues. Here Q(SASA) is defined as the quotient of the SASA (solvent-accessible surface area) and the surface area of the isolated residue, using the default probe radius of 1.4 Å. With the introduction of AlphaFold2 structural models to TITINdb2, we have also annotated domain-domain interface residues in domains; these reflect residues which appear surface-exposed when the single domain is assessed, but are buried when the adjacent domains are included in the model. To determine interface residues, tandem domains (domains separated by a linker of < 10 residues) were taken and the SASA of each residue was calculated. Residues were annotated as interface-buried if the SASA of the residue in the single-domain system decreased by at least 30 Å2 in the tandem-domain system (see Fig.5). This was repeated for all available fragments in which the domain tandem was represented, and residues confirmed as interface-buried if the majority of fragments were in agreement (> 50% interface-buried).
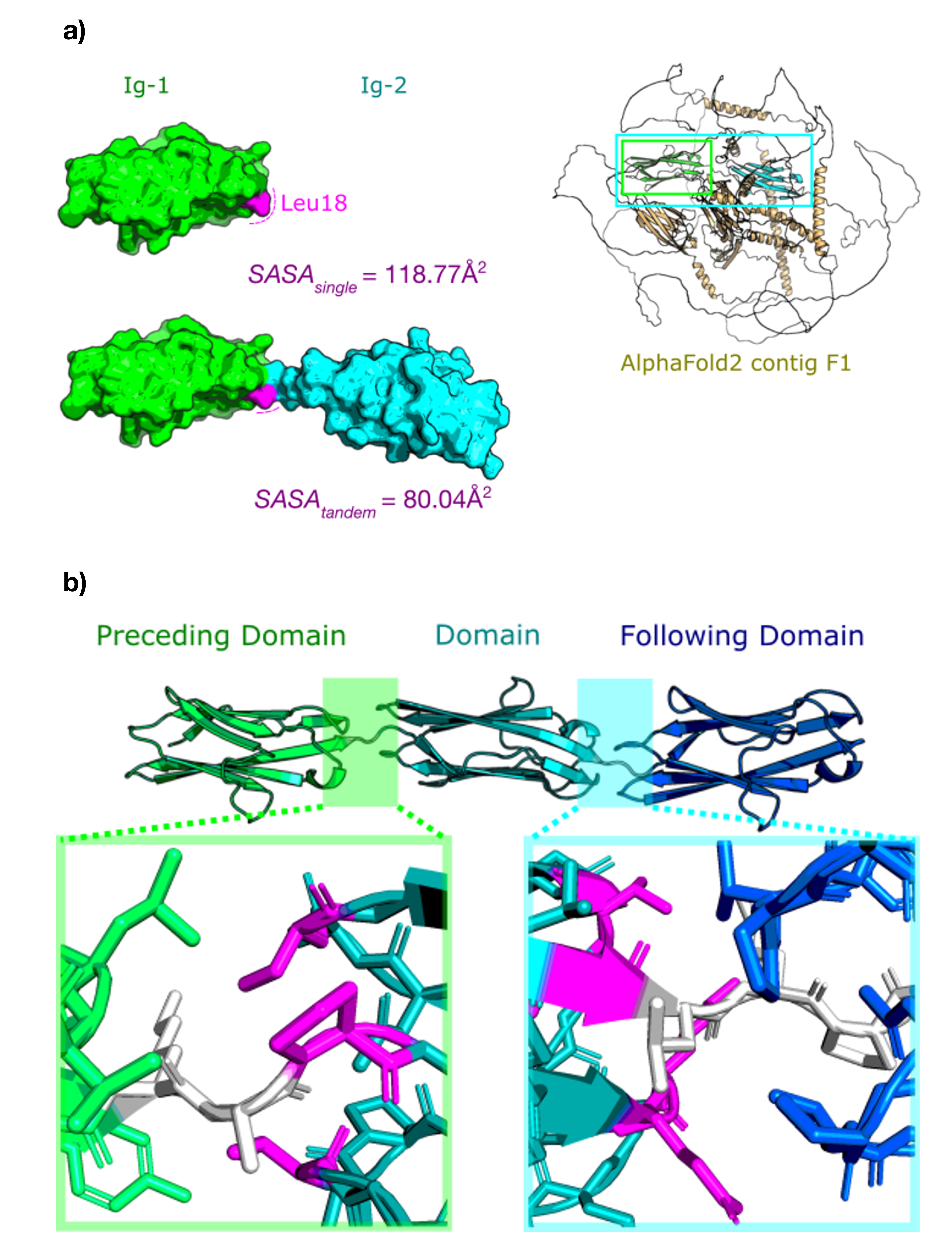
A second category of "boundary" was also defined, which incorporated residues defined as interface-buried within 10 residues of the N- or C-termini of the domain, in order to prevent residues in linkers or at the domain boundaries from being erroneously labelled as interface-buried.
Protein-Protein Interactions and Site Annotations
The assessment of the impact of SNVs on protein-protein interactions was performed using mCSM [15], where experimental binary complexes were available. The following structures were used for these calculations:
| Domain | PDB Structure | Solution Method | Resolution | Interacting Protein |
|---|---|---|---|---|
| Ig-1 | 1YA5 | X-ray Diffraction | 2.44Å | TCAP |
| Ig-2 | 1YA5 | X-ray Diffraction | 2.44Å | TCAP |
| Ig-169 | 3KNB | X-ray Diffraction | 1.40Å | OBSL1 |
| Ig-169 | 4C4K | X-ray Diffraction | 1.95Å | OBSCN |
Putative PPI interface regions were predicted using SPPIDER II [50] with a balanced trade-off between sensitivity and specificity (SPPIDER estimates this based on a control data set of 149 protein chains with no sequence homology).
Site annotations, including ligand binding sites and modified residues, were obtained from UniProt [51].
TITINdb2 Variant Converter
The titin variant conversion tool was developed to aid clinicians and researchers working with different standard formatting for titin variants to quickly and reliably translate variants between protein, transcript, and chromosomal positions in the seven major isoforms (IC, N2AB, N2B, N2A, novex-1, novex-2, novex-3) in bulk. In brief, by entering the query variants into the provided textbox (whitespace separated) and selecting the desired input and output formats, the tool will return a list consisting of the input variants translated to the output format, again whitespace separated.
The conversion from SAV to SNV relies on coercion of the wild-type codon to a mutant codon matching the mutated residue; if multiple codon substitutions match the amino acid substitution, the tool will only report the first match. Additionally, the tool only supports the conversion of SNVs and SAVs; it will not, for example, translate an SAV that corresponds to an indel at the genetic level (e.g. the SAV C31712R may be coerced to the SNV 95359T>C, but the theoretical SAV C31712K could not be coerced to the indel 95359_95360delinsAA). The format of input variants should follow the simple variant nomenclature syntax outlined below, and not the standard HGVS formatting for variants:
| Format (IC) | Example HGVS Nomenclature | TITINdb2 Variant Converter |
|---|---|---|
| Chromosome | NC_000002.12:g.178546102A>G | 178546102A>G |
| Transcript | NM_001267550.2:c.95359T>C | 95359T>C |
| Protein | NP_001254479:p.Cys31712Arg | C31712R |
In addition to selecting isoform and format by name, users may select input and output formats using accession IDs derived from RefSeq [36], UniProt [51] and Ensembl [52]. These resources were also used to cross-reference sequence data for protein and transcript isoforms. Data for genetic sequences, exon boundaries, and chromosomal numbering were derived from CardioDB [53][54].
Note that the 225bp upstream non-translated sequence is included in the numbering for all titin transcripts; thus, the ATG start codon for the initial methionine residue in all isoforms is numbered as 226-228. This may be inconsistent with other numbering schemes that omit these nucleotides, thus users should be aware of these potential differences.
Citing TITINdb2
When using this tool in publication, please cite: Weston, T., Ng, J., Gracia Carmona, O., Gautel, M., & Fraternali, F. (2025). "TITINdb2—expanding annotation and structural information for protein variants in the giant sarcomeric protein titin.". Bioinformatics Advances, 5(1)
To cite the literature review for disease-associated missense variants in titin: Weston, T., Rees, M., Gautel, M. & Fraternali, F. "Walking with giants: The challenges of variant impact assessment in the giant sarcomeric protein titin". WIREs Mechanisms of Disease, e1638.
References
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68-74 (2015).
- Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434-443 (2020).
- Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29, 308-311 (2001).
- The Genome Aggregation Database. https://gnomad.broadinstitute.org/
- UK Biobank - Enabling scientific discoveries that improve human health. https://www.ukbiobank.ac.uk/
- gnomAD v4.0 Announcement. https://gnomad.broadinstitute.org/news/2023-11-gnomad-v4-0/
- Chauveau, C., Rowell, J. & Ferreiro, A. A Rising Titan: TTN Review and Mutation Update. Human Mutation 35, 1046-1059 (2014).
- Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2018).
- Weston, T. G. R., Rees, M., Gautel, M. & Fraternali, F. Walking with giants: The challenges of variant impact assessment in the giant sarcomeric protein titin. WIREs Mechanisms of Disease, e1638.
- Fowler, D. M. & Fields, S. Deep mutational scanning: a new style of protein science. Nat Methods 11, 801-807 (2014).
- Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr Protoc Hum Genet 0 7, Unit7.20 (2013).
- Ponzoni, L., Peñaherrera, D. A., Oltvai, Z. N. & Bahar, I. Rhapsody: predicting the pathogenicity of human missense variants. Bioinformatics 36, 3084-3092 (2020).
- Pires, D. E. V., Ascher, D. B. & Blundell, T. L. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30, 335-342 (2014).
- Pires, D. E. V., Ascher, D. B. & Blundell, T. L. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res 42, W314-W319 (2014).
- González-Pérez, A. & López-Bigas, N. Improving the Assessment of the Outcome of Nonsynonymous SNVs with a Consensus Deleteriousness Score, Condel. Am J Hum Genet 88, 440-449 (2011).
- Ioannidis, N. M. et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet 99, 877-885 (2016).
- Functional ANnotations for Non-Synonymous SNVs. https://bbglab.irbbarcelona.org/fannsdb/
- Cheng, J. et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 0, eadg7492 (2023).
- Cheng, J. et al. Predictions for AlphaMissense. (2023) doi:10.5281/zenodo.8360242
- Liu, X., Li, C., Mou, C., Dong, Y. & Tu, Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Medicine 12, 103 (2020).
- Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47, D886-D894 (2019).
- Shihab, H. A. et al. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum Mutat 34, 57-65 (2013).
- Jagadeesh, K. A. et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet 48, 1581-1586 (2016).
- Dong, C. et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 24, 2125-2137 (2015).
- Samocha, K. E. et al. Regional missense constraint improves variant deleteriousness prediction. 148353 (2017) doi:10.1101/148353.
- Reva, B., Antipin, Y. & Sander, C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Research 39, e118 (2011).
- Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11, 361-362 (2014).
- Pejaver, V. et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat Commun 11, 5918 (2020).
- Qi, H. et al. MVP predicts the pathogenicity of missense variants by deep learning. Nat Commun 12, 510 (2021).
- Sundaram, L. et al. Predicting the clinical impact of human mutation with deep neural networks. Nat Genet 50, 1161-1170 (2018).
- Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLOS ONE 7, e46688 (2012).
- REVEL: Rare Exome Variant Ensemble Learner - REVEL genome segment files. https://sites.google.com/site/revelgenomics/downloads/revel-genome-segment-files .
- Ng, P. C. & Henikoff, S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 31, 3812-3814 (2003).
- Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583-589 (2021).
- Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39, W29-37 (2011).
- O'Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733-745 (2016).
- Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res 42, D222-230 (2014).
- Crooks, G. E., Hon, G., Chandonia, J.-M. & Brenner, S. E. WebLogo: a sequence logo generator. Genome Res 14, 1188-1190 (2004).
- Notredame, C., Higgins, D. G. & Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205-217 (2000).
- Myers, E. W. & Miller, W. Optimal alignments in linear space. Comput Appl Biosci 4, 11-17 (1988).
- Pei, J., Kim, B.-H. & Grishin, N. V. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res 36, 2295-2300 (2008).
- Armstrong, D. R. et al. PDBe: improved findability of macromolecular structure data in the PDB. Nucleic Acids Res 48, D335-D343 (2020).
- Eswar, N. et al. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics Chapter 5, Unit-5.6 (2006).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9, 40 (2008).
- AlphaFold Protein Structure Database. https://alphafold.ebi.ac.uk/
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 1.8. (2015).
- Williams, C. J. et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci 27, 293-315 (2018).
- Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat Methods 19, 679-682 (2022).
- Cavallo, L., Kleinjung, J. & Fraternali, F. POPS: A fast algorithm for solvent accessible surface areas at atomic and residue level. Nucleic Acids Res. 31, 3364-3366 (2003).
- Porollo, A. & Meller, J. Prediction-based fingerprints of protein-protein interactions. Proteins 66, 630-645 (2007).
- UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47, D506-D515 (2019).
- Martin, F. J. et al. Ensembl 2023. Nucleic Acids Research 51, D933–D941 (2023).
- Roberts, A. M. et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med 7, 270ra6 (2015).
- Titin Variation in Dilated Cardiomyopathy - Cardiovascular Genetics & Genomics, Imperial College London. https://www.cardiodb.org/titin/titin_transcripts.php.